Casos Clínicos
Manejo Odontoestomatológico del Síndrome de Costello: a propósito de un caso clínico
Recibido para Arbitraje: 16/07/2014
Aceptado para publicación: 13/03/2015Morales-Chávez, M.C., Profesor Agregado, Directora del Centro de Investigaciones de la Universidad Santa María. Caracas- Venezuela. Nualart-Grollmus, Z.C., Profesora Asistente de la Universidad Mayor, Temuco, Chile. Silvestre-Donat, F.J., Profesor Titular de Medicina Oral y Pacientes Especiales, Universidad de Valencia - España.
CORRESPONDENCIA: [email protected]
MANEJO ODONTOESTOMATOLÓGICO DEL SÍNDROME DE COSTELLO: A PROPÓSITO DE UN CASO CLÍNICO
RESUMEN
El Síndrome de Costello (SC) conocido como Síndrome faciocutaneoesquelético es una enfermedad congénita de origen genético causado por una mutación heterocigota en la línea germinal del gen HRAS, localizado en el cromosoma 11p15.5 en el 85% de los casos, caracterizado por retraso en el crecimiento, retraso mental, rasgos faciales toscos, alteraciones del sistema estomatognático, cardiopatías; así como tendencia a desarrollar tumoraciones benignas y malignas, muchas de las cuales ocurren en la cavidad bucal y sus zonas adyacentes. Los casos que dan resultados negativos para mutaciones en HRAS podrían ser explicados por la presencia de la heterogenicidad de locus, es decir la presencia de otros genes en diferentes regiones cromosómicas, cuya alteración sea causante del síndrome. Debido a la complejidad del síndrome y a la necesidad de tomar consideraciones especiales a la hora de realizar tratamiento odontológico ya que solo se encuentran reportados en la literatura cerca de un centenar de casos, se describirá un caso clínico.
PALABRAS CLAVE: Síndrome de Costello, síndrome faciocutáneoesqueletal, retardo mental, rasgos faciales toscos.
ODONTOSTOMATOLOGY MANAGEMENT OF COSTELLO SYNDROME: A CASE REPORT
ABSTRACT
Costello Syndrome (CS) is a congenital disease of genetic origin caused by heterozygous germline HRAS mutation in the chromosome 11p15.5 in the 85% of the cases, which is characterized by slow developmental growth (both physical and mental), coarse face, stomatognathic system alterations, and cardiac problems. Furthermore, this disease also tends to result in the development of benign and malignant tumors, much of these on the oral and perioral area. Cases that are negative for HRAS mutations may be explained by the presence of locus heterogeneity, like the presence of other genes in different chromosomal regions, which is cause of the alterations. This article describes a CS clinical case, looking to increase the number of reported cases in the literature of this complex syndrome. The article will focus on the special considerations needed to be taken before starting the oral treatment.
KEY WORDS: Costello Syndrome, Facio-cutaneous-skeletal syndrome, mental retardation, coarse face. |
Costello, de Nueva Zelanda, describió en una reunión científica en 1977 dos casos de niños con retardo mental, alto peso al nacer, problemas de alimentación en el período neonatal, cabello enrollado, facies toscas y nariz bulbosa y papilomatosa, síndrome que tiempo después recibiría su nombre
1. Años después se determinó que la entidad descrita con el nombre de síndrome Facio-cutáneo-esqueletal y el síndrome de Costello eran la misma. Posteriormente el SC fue diagnosticado en muchos otros pacientes y recibió aún más atención cuando la tendencia incrementada de desarrollo de lesiones malignas se esclareció
2. Esta enfermedad congénita, tiene un patrón de herencia autosómica dominante, con penetrancia completa y expresividad variable
1-9. EL primer reporte de mutaciones génicas causantes de SC se publicó en el año 2005. Debido al solapamiento fenotípico del SC con el síndrome de Noonan, causado por mutaciones en el gen PTPN11, se analizaron diferentes genes de la misma ruta metabólica. El resultado fue la identificación de 12 mutaciones heterocigóticas del gen HRAS, localizado en el cromosoma 11p15.5. Las mutaciones en el gen HRAS han sido detectadas en el 85% de los pacientes con SC. Los casos que dan resultados negativos para mutaciones en HRAS podrían ser explicados por la presencia de la heterogenicidad de locus, es decir la presencia de otros genes en diferentes regiones cromosómicas, cuya alteración sea causante del síndrome
10,11.
En la actualidad hay 11 genes disponibles y de validez clínica responsables del funcionamiento de la ruta metabólica RAS/MAPK los que están asociados a los Síndromes de Noonan, Costello y Cariofaciocutáneo. Los genes son: PTPN11, SOS1, RAF1, KRAS, NRAS, CBL, BRAF, MEK1, MEK2, SHOC2, yHRAS, y es posible estudiarlos simultáneamente a través de un panel de estudios genéticos de alta sensibilidad técnica
10,12.
Los pacientes con SC tienden a nacer con un peso alto, que en 89% de los casos sobrepasa el percentil 50, alcanzando generalmente los cuatro kilos. Sin embargo, durante los primeros días de vida, el neonato perderá peso y desarrollará una incapacidad para su alimentación
1,5,6. Todo esto conlleva a que estos pacientes desarrollen un importante retraso en el crecimiento, asociado generalmente a un déficit en la hormona de crecimiento. Aunado a esto, manifiestan un retraso psicomotriz, así como un retardo mental de leve a severo en el 100% de los pacientes
2,3,4,8.
Sus principales diagnósticos diferenciales son el síndrome de Noonan y el síndrome cardio-facio-cutáneo
11. En edades tempranas estos tres síndromes pueden ser difíciles de diagnosticar. Posteriormente las diferencias con el síndrome de Noonan se hacen más evidentes. Los síndromes de Beckwith-Wiedemann y Simpson-Golabi-Behmel deben considerarse en el diagnóstico diferencias debido a la presencia neonatal de macrosomía, macroglosia, fascies tosca e hipoglucemia. El síndrome de Williams comparte con el SC la hiperlaxitus ligamentosa, piel redundante, labios prominentes. Sin embargo, no tiene un perfil cognitivo específico, arteriopatía, alteraciones de tejido conectivo y frecuentemente hipercalcemia
10.
Los signos faciales más comunes que caracterizan a un paciente con este síndrome son una absoluta o relativa macrocefalia, hirsutismo en la frente, una frente amplia, una nariz ancha con depresión del puente nasal, estrabismo, boca grande, labios muy gruesos e implantación baja de las orejas
2,4.
La combinación de macrocefalia con nariz bulbosa, boca grande, labios delgados y macroglosia hacen por lo general muy característicos a este síndrome. Sin embargo, algunos pacientes inicialmente fueron confundidos con el diagnóstico de síndrome de Hurley y otras mucopolisacaridosis
2.
A nivel intra bucal, suelen presentar hiperplasia gingival, macroglosia y paladar profundo. Algunos pacientes también pueden presentar úvula bífida. En relación a los dientes, suelen presentar muchos espacios entre ellos y presenta una prevalencia elevada de caries
2.
El sistema musculoesquelético también se encuentra afectado, presentando en la mayoría de los casos una hipermovilidad articular. Las manifestaciones dermatológicas del SC incluyen piel suave con arrugas en el dorso de las manos, hiperqueratosis en las plantas de los pies e hiperextensibilidad de los dedos. También presentan hiperpigmentación generalizada, los nevus y otras lesiones pigmentadas aparecen con frecuencia en las palmas de las manos, plantas de los pies y otras partes del cuerpo
2,3,4,13,14.
Un 80% de los pacientes con SC presentan cardiopatías congénitas. Estas pueden ser defectos anatómicos o anomalías estructurales como estenosis de la válvula pulmonar o aórtica, y alteraciones del septum ventricular. También pueden manifestarse
cardiomiopatía hipertrófica y arritmias
1-4,10,15.
Otra de las características distintivas del SC es la predisposición a tumores. Los tumores de origen ectodérmico como los epiteliomas calcificados, quistes dermoides, fibroadenosis mamaria o mastopatía fibroquística y papilomas. Los papilomas son muy comunes durante la niñez, desarrollándose principalmente en la zona perioral y perianal. Estos constituyen la patología más prevalente
13. Sin embargo, también han sido reportados el desarrollo de neurilemomas, rabdomiosarcomas, neuroblastomas y carcinomas de vejiga
2,5,6.
La incidencia de tumores en pacientes con SC puede compararse con el 7-21% que aparecen en el síndrome de Beckwith-Wiedemann. Sin embargo, los tipos de tumores son diferentes. En el caso de los rabdomiosarcomas, se considera el tumor maligno más frecuente en el SC
16.
Las proteínas de la vía RAS/MAPK desempeñan un papel fundamental en la proliferación, diferenciación, supervivencia y muerte celular. Desde hace más de 30 años se sabe que el 30% de los cánceres presentan una mutación somática en algunos de los genes que codifican estas proteínas. En contraste con el elevado potencial de malignidad de las mutaciones somáticas, las mutaciones en la línea germinal provocan anomalías en el desarrollo del individuo. Entre los síndromes conocidos como rasopatías se encuentran el síndrome de Costello, el síndrome de Noonan, la neurofibromatosis1, el síndrome de Leopard, el síndrome cardio-facio-cutáneo y el síndrome de Legius
17.
Se ha demostrado además que existe una posible relación entre los oncogenes de señalización HRAS y el balance del condroitin sulfato (CS) en el síndrome de Costello. Los Glucosaminoglicanos, incluyendo al Condroitin Sulfato están envueltos en una gran variedad de procesos biológicos. El balance de la síntesis de CS es controlada a través de expresiones temporales de distintos genes condrointin sulfotransferasas. Sin embargo, el balance puede verse alterado en algunas enfermedades. El CHST11/C4ST-1, después llamado C4ST-1 cataliza la transferencia de sulfato a la posición C-4 de los CS disacáridas, formándose Condroitin-4-sulfatato (C4S) como uno de sus productos. Se describió recientemente un balance alterado en la síntesis de CS con un déficit de los niveles de C4S en el tejido cardíaco de los pacientes con síndrome de Costello
18.
Estudios recientes concluyen que existe una estrecha relación en la expresión de C4ST-1 y el balance de sulfatación del Condroitin en la mediación de señalizadores HRAS en la patogénesis del síndrome de Costello. Específicamente se plantea que la reducción en la mediación HRAS de la expresión de C4ST-1 es necesaria para la aparición de las características fenotípicas del síndrome de Costello como anormalidades en la piel donde se incluyen los defectos en la elastogénesis. Igualmente se ha determinado que la reducción en la expresión de C4ST-1 es un pre requisito para varias de las anomalías observadas en el síndrome, tales como defectos cardíacos y la formación de neoplasias
18.
Estudios realizados en ratones han mostrado evidencia de que el gen HRAS puede estar involucrado en la iniciación de la carcinogénesis. Algunos de estos estudios se han realizado en tumoraciones en la piel de ratones, demostrándose que son causados por una transición específica en el codón 12 del gen HRAS
15.
Se ha demostrado recientemente una relación entre el SC y la enfermedad de Alzheimer. Ambas condiciones son la expresión de enfermedades genéticas. Cuando hay defecto en un solo gen es un ejemplo de Alzheimer pero cuando hay mutaciones en varios genes de los elementos de la vía de señalización, pueden generarse condiciones como el síndrome de Costello
19.
CASO CLÍNICOSe trata de una mujer de 20 años de edad, que acude al Servicio de Odontología para Pacientes con Discapacidad de la Cruz Roja en la ciudad de Valencia, España para se atendida.
Al interrogar a la representante sobre los antecedentes médicos de la paciente, ésta refiere que ha presentado una Angiodisplasia de Colon con antecedentes de sangrado, encontrándose en ese momento bajo tratamiento farmacológico con anticoagulantes. Igualmente refiere un derrame pleural derecho y una miocardiopatía controlada, consistente en una comunicación interauricular y displasia de la válvula mitral y tricúspide. Finalmente la madre hace referencia a que a la paciente tiene diagnóstico de Síndrome de Costello concomitante con retraso mental moderado.
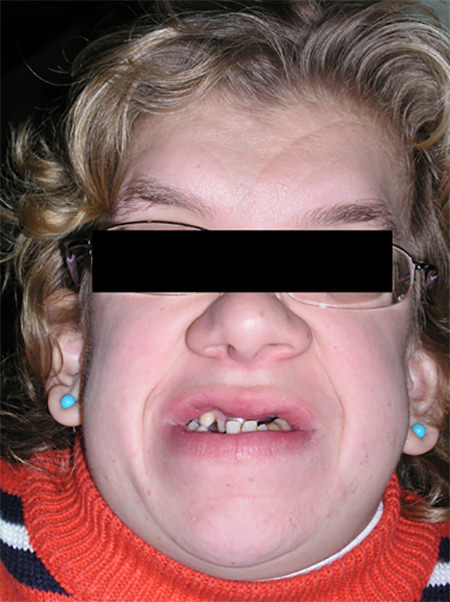
Posterior a la recolección de datos en la historia clínica y a la firma por parte la de representante del consentimiento informado, cumpliendo con las normas éticas de la institución, se procedió a realizar el examen clínico. Al examen físico se determinó una estatura de 1,38 m, coincidiendo con el percentil 3 según Hernández y cols. En el examen extraoral se observaron los rasgos faciales típicos de síndrome como frente amplia, nariz ancha, cabello rizado, asimetría de los tres tercios de la cara, siendo más amplio el tercio inferior (Fig 1).
 |
| Fig 1. |
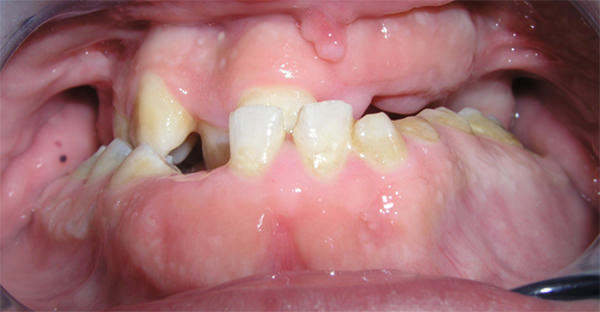
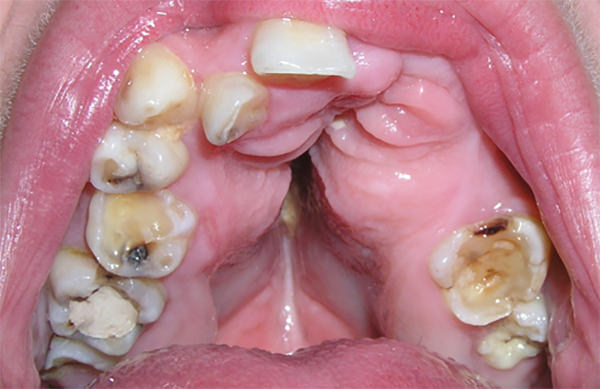
Dentro de la cavidad bucal de la paciente se observó que la mandíbula sobrepasa al maxilar, generándose una mordida cruzada anterior y posterior. Así mismo se determinó un colapso del maxilar, fisura palatina submucosa, macroglosia, ausencias dentarias, caries y atriciones dentarias, así como gingivitis asociada a placa dental. En la mucosa de la cara interna del carrillo del lado derecho se diagnosticó la presencia de una mácula pigmentada de 1 mm de diámetro y una lesión con diagnóstico presuntivo de fibroma a nivel del frenillo labial (Fig 2 y 3).
 |
| Fig 2. |
 |
| Fig 3 |
El tratamiento de esta paciente se llevó a cabo en el servicio, sin necesidad de recurrir a anestesia general o sedación ya que el retardo mental de la paciente permitió acondicionarla a la consulta odontológica. Se realizaron las restauraciones correspondientes y el tratamiento peridontal. Así mismo la paciente fue referida al Servicio de Medicina Bucal para la eliminación de la lesión localizada a nivel del frenillo labial, donde se realizó la extirpación de la misma y posterior biopsia, dando como resultado un fibroma. Igualmente fue enviada al Servicio de Ortodoncia para evaluar la posibilidad de tratamiento.
DISCUSIÓN:El SC fue descrito por Costello en 1977, estos pacientes presentan un cuadro complejo y multisistémico, dentro del cual se encuentra inmerso el sistema estomatognático
1-6. El síndrome de Costello forma parte de un grupo de síndromes de desarrollo llamados RASopatías, las cuales son causadas por mutaciones de línea germinal en los genes que codifican los componentes proteicos de las vías de señalización celular RAS / mitogen de la proteína quinasa activada (MAPK). La vía de RAS 7 MAPK es importante en la regulación del ciclo celular, la diferenciación y el crecimiento celular
17,19.
En el caso descrito, la paciente presenta una cardiopatía como en el 80% de los pacientes con SC que presentan cardiopatías asociadas. Por esta razón, esta condición debe ser tomada en consideración por el odontólogo, haciendo interconsulta con el médico tratante y entre ambos determinar la necesidad de una profilaxis antibiótica para ciertos tratamientos odontológicos
2-5.
Respecto al retraso mental que se presenta en el 100% de los pacientes, se debe asumir que esto traerá consecuencias en el estado de salud bucodental ya que los pacientes con déficit cognitivos, tienen menor capacidad para realizar la higiene bucal. Igualmente, el 20% tiende a desarrollar convulsiones, por lo que se hace necesario indicar una terapia farmacológica anticonvulsivante que en el 36 - 67% de los casos desarrollará hiperplasia gingival
2,4,21-23.
La dificultad que presentan estos pacientes para aprender a masticar y deglutir creará en la mayoría de estos niños problemas severos en la oclusión como es el caso de la paciente descrita que tiene una mordida cruzada anterior y posterior
2,4,6.
Otra consideración importante a tomar en cuenta es que debido al retraso mental, muchas veces severo se hará imposible la atención en el consultorio odontológico por lo cual el paciente será referido a sedación o anestesia general. La implicación de múltiples sistemas en el SC, así como la vía aérea y las afecciones cardíacas juegan un rol muy importante en los cuidados perioperatorios. Ciertas de las características de estos pacientes como el cuello corto, macroglosia, hipertrofia amigdalina y de los tejidos supraglóticos, la presencia de papilomas laríngeos, así como la atresia de las coanas, crean verdaderas dificultades para el anestesiólogo a la hora de entubar a un paciente con este síndrome, por lo cual la remisión a anestesia general es un tratamiento que debe dejarse solamente para casos de emergencia, e intentar siempre, el acondicionamiento bajo técnicas psicológicas, evitando de esta manera serias complicaciones médicas
8,9,24.
En 2013 Takahashi y cols evaluaron a cuatro pacientes con síndrome de Costello mediante tomografías computadorizadas. Los cuatro pacientes presentaron una macrocefalia con hipoplasia ósea facial e hipertrofia gingival. Así mismo se observaron atriciones dentales, maloclusiones y arcos maxilares pequeños al igual que en el paciente descrito. Además, un paciente presentó caries dental, dientes cónicos, gingivitis asociada a placa dental e hipodoncia
25.
CONCLUSIONES:La tarea del odontólogo se basa en brindar salud y calidad de vida a los pacientes. Sin embargo, debe considerarse que muchos de ellos presentan discapacidades físicas, mentales o psicológicas que interfieren con su crecimiento y desarrollo normal y que el especialista debe enfrentar para lograr una salud integral.
Realmente los odontólogos deben capacitarse para la odontología del nuevo milenio, esa que se dedica a la atención de pacientes con discapacidad y médicamente comprometidos, la verdadera odontología del futuro.
REFERENCIAS BIBLIOGRÁFICAS:
- Costello JM: A new syndrome: Mental subnormality and nasal papillomata. Aust Pediatr J (1977); 13:114-8.
- Hennekam R: Costello Syndrome: An overview. Am J Med Genet Part C (2003); 117C: 42-8
- Pascual-Castroviejo I, Pascual-Pascual S.I: Síndrome de Costello. Presentación de un caso con seguimiento durante 35 años. Neurología. (2005); 20 (3): 144 -8.
- Van Eehen A, Van Gelderen I, Hennekam R: Costello Syndrome: Report and Review. Am J Med Genet. (1999); 82: 187 - 93.
- White SM, Graham J.M, Kerr B; Gripp K; Weksberg R, Cytrynbaum C, Reeder JL, Stewart FJ, Edwards M, Wilson M, Bankier A: The Adult Phenotype in Costello Syndrome. Am J Med Genet. (2005); 136A: 128-35
- Gripp K. Tumor Predisposition in Costello Syndrome. Am J Med Genet. (2005); 137C: 72-7.
- Katcher K, Bothwell M, Tobias J: Case Report. Anaesthetic implications of Costelo Syndrome. PAEDIATR ANAESTH. (2003); 13: 257-62.
- Della Marca G, Vasta I, Scarano E, Rigante M, De Feo E, Mariotti P, Rubino M, Vollono C, Mennuni GF, Tonali P, Zampino G: Obstructive Apnea in Costello Syndrome. Am J Med Genet. (2006); 140A: 257-62.
- Saji A, Ramadan D, Alkhayyant H, Al-Sharkawi I, Aboo KC, El-Sabban F, Hussain K: Costello Syndrome and Hyperinsulinemic Hypoglycemia. Am J Med Genet. (2005); 139A: 227-30.
- Martínez-Glez V, Lapunzina P. Síndrome de Costello. Grupo de Trabajo sobre Cáncer en Síndromes Genéticos Polimalformativos (GT-CSGP). obtenible en: < http://www.orpha.net/data/patho/Pro/es/CSGP-Costello-3.pdf > [consulta : 21 marzo 2014]
- Figueroa H, Covarrubias N, Santander D, González A, Bravo D, Urbina M, Barra R. Síndrome de Costello: Reporte de un Caso. In proceeding XLIX Congreso Chileno de Pediatría. (2009). Obtenible en < http://www.researchgate.net/publication/ 210164788_SNDROME_DE_COSTELLO._REPORTE_DE_UN_CASO > [Consulta:_ 26 marzo 2014]
- Puppio A. Breve descripción de la vía Metabólicaras/MAPK y genes responsables asociados a síndrome de Noonan, Costello y Cardiofaciocutáneo. (2012). Obtenible en: http://es.scribd.com/doc/79675727/Sindrome-de-Noonan-Cardio-Facio-Cutaneo-Costello-Genes-responsables-de-la-via-metabolica-RAS-MAPK [Consulta: 25 marzo 2014].
- Nguyen V. Buka RL, Roberts BJ, Eichenfield LF. Cutaneous manifestations of Costello syndrome. Int J Dermatol. (2007) Jan;46(1):72-6.
- Aytekin S, Alyamac G: Two new cases with Costello syndrome. Dermatol Online J. (2013); Aug 15;19(8):19267.
- Estep AL, Tidyman WE, Teitell MA, Cotter PD, Rauen KA. HRAS mutations in Costello syndrome: detection of constitutional activating mutations in codon 12 and 13 and loss of wild-type allele in malignancy. Am J Med Genet A. 2006;140:8-16.
- Gripp KW, Scott CI Jr, Nicholson L, MacDonald-McGinn DM, Ozeran JD, Jones MC, Lin AE, Zachai EH. Five additional Costello síndrome patientes with rhabdomyosarcoma:proposal for a tumor screening protocol. Am J Medi Genet. (2002) 108(1):80-7.
- Hernández-Martin A, Torrelo-Fernández A. Rasopatías: trastornos del desarrollo con predisposición al cáncer y manifestaciones cutáneas. Actas Dermo-sifiliográficas. (2011) 102(6): 402-16.
- Klüppel M, Samavarchi-Tehrani P, Liu K, Wrana JL, Hinek A: C4ST-1/CHST11-controlled condroitin sulfatation interferes with oncogenic HRAS signaling in Costello syndrome. Eur J Hum Genet. (2012) 20, 870-7.
- Cie?lewicz AR1, Jab?ecka A. Genetic base of heart diseases. Pol Merkur Lekarski. (2010) Jul;29(169):5-7.
- Tajir M, Fergelot P, Lancelot G, Arveiler B, Elelaoui SC, Lacombe D, Sefiani A: Syndrome de Costello: à propos d'une observation. Pan Afr Med J. (2012) 12:64.
- Roldán S, Herrera D: Tratamiento, Mantenimiento y Prevención en Periodoncia. Barcelona. Dentaid. 2005.
- Bullón P, Machuca G: Tratamiento Odontológico en Pacientes Especiales. Segunda Edición.Madrid. Laboratorios Normon, S.A. 2004.
- Morales-Chávez MC: Odontología en Pacientes Especiales: Una Necesidad Creciente. Caracas. Editorial Colson. 2012.
- Kopel, HM: The Autistic Child in Dental Practice. ASDC J Dent Child. Jul-Aug. (1997) 44(4): 302-9.
- Takahashi M, Ohashi H: Craniofacial and dental malformations In Costello syndrome: A detailed evaluation using multi-detector row computed tomography. Congenit Anom (Kyoto). (2013); Jun;53(2):67-72.
|
