Síndrome de Albright-Mccune Sternberg: reporte de un caso y revisión de la literatura
Recibido para Arbitraje: 13 /09/2013
Aceptado para Publicación: 13/11/2013
López, B., López, E., López Heriberto, R., López, M., Odontólogos egresados de la Facultad de Odontología de la Universidad de Carabobo, Venezuela. López Heriberto, J., Profesor agregado de la Cátedra de Cirugía Bucal de la Facultad de Odontología de la Universidad de Carabobo, Venezuela. Mora, O. Profesor titular de la Cátedra de Cirugía Bucal de la Facultad de Odontología de la Universidad de Carabobo, Venezuela.
SÍNDROME DE ALBRIGHT-McCUNE STERNBERG: REPORTE DE UN CASO Y REVISIÓN DE LA LITERATURA
RESUMEN
El síndrome de Albright-McCune Sternberg (SAMS) es un desorden raro que se origina de una mutación del gen GNAS1. Se caracteriza por presentar un fenotipo típico, el cual incluye fibrodisplasia (FD) poliostótica, pubertad precoz (PP), pigmentaciones café au lait (café con leche) junto con otras endocrinopatías. La presente investigación trata de una paciente femenina de 22 años de edad con SAMS la cual presenta algunos signos y síntomas del síndrome tales como: FD poliostótica, pigmentaciones color café con leche en la piel, PP e hipotiroidismo. Acudió por dolor a nivel de las encías inferiores producto de un aumento óseo bimaxilar, inicia su enfermedad actual en mayo de 2013 presentando dolor a nivel de mucosa gingival inferior, localizado, punzante, de intensidad moderada, el cual se agrava ante la masticación y dura hasta el cese del estimulo. La FD fue diagnosticada posterior a la realización de una biopsia de tejido óseo y estudios radiográficos, la paciente presentó metrorragia a los nueve meses de edad el cual se repitió a los cinco años y persistió de manera intermitente hasta los veinte años de edad donde fue diagnosticada con ovarios poliquísticos por lo cual se le prescribió etinilestradiol y acetato de ciproterona. Aunque el SAMS generalmente cursa con una hiperfunción endocrina la paciente tiene un diagnóstico de hipotiroidismo por lo cual está bajo tratamiento con levotiroxina.
ALBRIGHT-McCUNE STERNBERG SYNDROME: CASE REPORT AND LITERATURE REVIEW
ABSTRACT
The Albright-McCune Sternberg syndrome (AMSS) is a rare disorder that arises from a mutation of the GNAS1 gene. It is characterized by a typical phenotype, which includes polyostotic fibrous dysplasia (FD), precocious puberty (PP), cafe-au-lait pigmentations and other endocrinopathies. The following research is about a 22 year old female patient with SAMS which presents some signs and symptoms of the syndrome such as polyostotic FD, pigmentation of the skin, PP and hypothyroidism. She attended by pain in the lower gum product of increased bimaxillary volume it began in may 2013 having pain in lower gingiva localized, throbbing, of moderate intensity, which is exacerbated by chewing and lasts until the cessation of the stimulus. The FD was diagnosed after performing a bone biopsy and radiographic studies, the patient had metrorrhagia at nine months of age which was repeated at five years and persisted intermittently until twenty years where he was diagnosed with polycystic ovaries so was prescribed ethinylestradiol and cyproterone acetate. Although the AMSS usually occurs with endocrine hyperfunction the patient has a diagnosis of hypothyroidism which is treated with levothyroxine.
El Síndrome de Albright-McCune Sternberg (SAMS) es un trastorno esporádico, complejo, no hereditario, originalmente definido por la triada clínica de FD ósea poliostótica, pigmentaciones cutáneas color café con leche y PP periférica 1. El diagnostico se considera confirmado cuando por lo menos dos de estos signos cardinales están presentes. El fenotipo del SAMS es causado por la activación de una mutación somática postcigotica del tipo mosaicismo, del gen GNAS1 con diferentes manifestaciones clínicas dependiendo del tejido que contenga dicha mutación 2. Normalmente se reemplaza la codificación normal de arginina por histidina (R201H), cisteína (R201C), leucina (R201L), o serina (R201S) en la subunidad "a" de la proteína Gs (Gas) en el codón 201 del gen, responsable de codificar la subunidad alfa de la proteína G heterotrimerica transductora de señal 3. El resultado es la activación constitutiva de la enzima adenilatociclasa y una sobreproducción de la 3´5´adenosin monofosfato cíclico 4. Posteriormente se reconocieron otras endocrinopatías asociadas al SAMS, dentro de las cuales se incluyen: hipertiroidismo, exceso de hormona del crecimiento, pérdida renal de fosfato con o sin raquitismo/osteomalacia y se puede encontrar síndrome de Cushing en asociación con la triada original. Rara vez otros sistemas de órganos pueden estar involucrados (hígado, corazón, paratiroides, páncreas) 5. Mientras que el SAMS es raro la FD no lo es, la FD puede involucrar un solo sitio esquelético o afectando varios a la vez. La PP puede ser encontrada asociada a las maculas cutáneas café con leche en ausencia de FD (cerca del 1% de los casos), pero en general, la FD es el componente más común del SAMS. Por esto una definición más amplia que la triada original de FD+PP+café con leche es SAMS=FD+ por lo menos una de las endocrinopatías hiperfuncionales típicas y/o maculas café con leche, con casi cualquier combinación posible 5.
La prevalencia del síndrome se ha reportado entre 1/100000-1/1000000 afectando con más frecuencia a las mujeres. Este síndrome fue descrito por primera vez por Fuller Albright en Massachusetts y por Donaban McCune en la Universidad de Columbia 1936. A través del tiempo varios reportes han llevado a un mejor entendimiento del síndrome y su fisiopatología 2. En el año 2001 en la revista médica de chile, se realizó una investigación bajo el título de "Estudio clínico-molecular de pacientes chilenas con síndrome de McCune-Albright" donde se estudiaron 14 niñas con dos o más manifestaciones clínicas sugerentes de SAMS y 33 controles sanos sin manchas café con leche en la piel, ni antecedentes de pubertad precoz o fracturas patológicas. Entre el grupo control 15 eran niñas < 7 años, 6 eran adolescentes (4 niñas y 2 varones) y 12 eran adultos jóvenes (10 varones y 2 mujeres). Para el estudio se tomó una muestra de ADN bajo el consentimiento previo de los padres los pacientes. El ADN genómico de pacientes y controles fue obtenido de leucocitos sanguíneos por el método de salting out donde fueron doblemente investigadas para la mutación R201H por digestión enzimática y por reacción de polimerasa en cadena alelo específica (PCR-AE). En los resultados se pudo señalar que todas las muestras obtenidas de los controles, amplificaron para el gen normal. Por otro lado, en 10 de los 14 pacientes con características clínicas propias de este síndrome se confirmó la presencia de la mutación mediante digestión enzimática 6.
CARACTERÍSTICAS CLÍNICAS
La clínica que este síndrome presenta es esencial para su correcto diagnóstico. El fenotipo completo del SAMS incluye FD ósea poliostótica, pigmentaciones café con leche y una hiperfunción endocrina 1. La FD es una lesión ósea benigna que está caracterizada por un reemplazo de hueso normal por una mezcla de tejido fibroso y pequeños fragmentos de hueso trabecular inmaduro. La lesión se presenta en el esqueleto en crecimiento con deformidades angulares que son el resultado de la mineralización defectuosa de la porción displásica en el interior de los huesos inmaduros. Al ser una afectación primaria mesenquimática, responsable de la formación del hueso, el tejido fibroso se expande y progresivamente reemplaza al hueso 7.
Al afectar varios huesos la displasia es conocida como poliostótica y al estar presente en un solo hueso es monostótica, esta última ha presentado una incidencia de 75 % a 80 % de los casos, de lo cual 20 % afecta los huesos craneofaciales, sin embargo, pueden comprometer múltiples huesos contiguos del cráneo, sin respetar las líneas de sutura. En la periferia normalmente afecta al fémur, especialmente al cuello, la tibia, las costillas, desarrollándose en la parte central del hueso. Generalmente está separada del cartílago de crecimiento en los niños y de la superficie articular en los adultos 8. En el tipo poliostótico la distribución y extensión de las lesiones varía ampliamente, desde el compromiso de unos pocos huesos de una extremidad hasta la afectación de más del 50% de los huesos del esqueleto, 90% de los casos es unilateral. Frecuentemente está implicada la pelvis, seguida de los huesos largos, cráneo, costillas, y extremidad proximal del fémur. Las lesiones poliostóticas tienden a permanecer más activas o agresivas. Se han descrito algunos casos donde la lesión sufre una transformación maligna a osteosarcoma o fibrosarcoma. A diferencia de la forma monostótica, la displasia fibrosa poliostótica da síntomas. Los hallazgos incluyen dolor, fractura espontanea, dificultad para caminar, o deformidad del miembro afectado 7. La apariencia radiográfica de las lesiones no es constante y depende del componente óseo y fibroso presente. Si predomina el componente de tejido conectivo se observará una lesión diafisiaria intramedular radiolúcida junto con un adelgazamiento de las corticales óseas. En cambio en el componente óseo se observa una lesión con aspecto de vidrio esmerilado o nebuloso que puede asociarse con desviaciones laterales o mediales con respecto al eje del hueso largo afectado 8.
Las manchas café con leche presentes en este síndrome son hiperpigmentaciones cutáneas mayores de 1 cm que generalmente aparecen en la infancia y tienden a incrementar su número y tamaño hasta la pubertad. Su color varía en distintos tonos de marrón y se localizan en cualquier parte del cuerpo independientemente de la exposición al sol, generalmente se concentran más en cara, cuero cabelludo, palmas de las manos y plantas de los pies y en los genitales externos 9. Los melanocitos cultivados a partir de estas lesiones muestran un aumento en los niveles intracelulares de AMPc, un mayor número de dendritas y melanosomas, y aumento de los niveles de la tirosinasa, la enzima limitante de la velocidad para la producción de melanina 10.
La hiperfunción endocrina característica de este síndrome produce una serie de manifestaciones clínicas importantes como parte del criterio diagnóstico, esta puede afectar una sola glándula o a múltiples de ellas. La más común entre ellas es la PP la cual es definida como un desarrollo de los cambios puberales antes de los 8 años producto de una incremento de la gonadotropina y/o los esteroides sexuales con un acelerado desarrollo somático de la edad ósea. Existen dos tipos de PP, aquella producto de una hiperfunción gonadal autónoma (PP periférica), aunque puede desarrollarse posteriormente hiperfunción hipofisaria (PP central). Aunque la PP en estos pacientes puede presentarse de manera intermitente y con largos períodos de remisión, algunos desarrollan una progresión rápida, con aceleración del crecimiento y la maduración esquelética, que comprometen la talla final. En relación a los signos clínicos observamos en hombres; vello púbico o alargamiento de genitales por debajo o entre los 7 y 10 años de edad, desarrollo de mamas antes de la aparición de vello púbico y del alargamiento de testículos; en las mujeres vello púbico antes de los 8 años y desarrollo de mamas por debajo de los 7 años, menarquia antes de los 9 a 11 años 9. El hipertiroidismo es común, se encuentra en 30 a 50% de los casos, aunque algunos pacientes presentan enfermedad subclínica. Independientemente de la presencia de hipertiroidismo clínico, en pacientes con SAMS existe elevación en la relación T3-T4, debida a un aumento de la actividad de las desyodinasas D1 y D2 mediada por la producción constitutiva de adenosínmonofostato. La producción excesiva de la hormona del crecimiento se encuentra en alrededor de 20% de los pacientes con SAMS aunque sólo algunos de ellos presentan un adenoma hipo?siario detectable por estudios de imagen. El exceso de hormona del crecimiento puede empeorar las lesiones craneofaciales, con disminución de la agudeza visual y auditiva, además de que ha sido implicado en la transformación sarcomatosa de las lesiones por Displasia Fibrosa. Éste es uno de los cuatro síndromes genéticos que son causantes de acromegalia 12.
REPORTE DE CASO
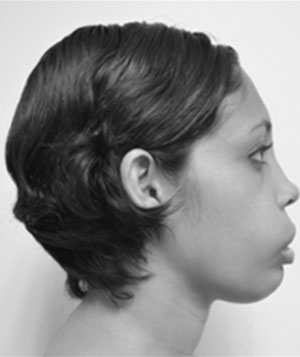
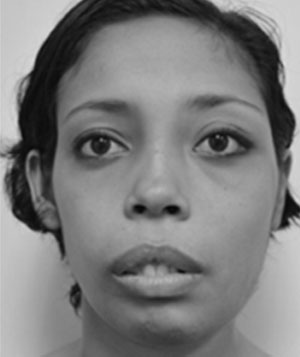
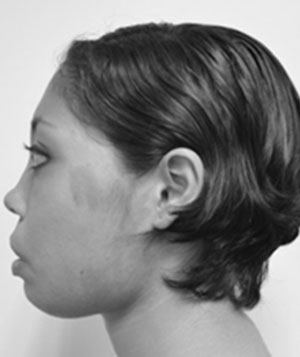
Se trata de paciente femenino de 22 años de edad, quien acude a la consulta por presentar dolor en la mucosa gingival de la mandíbula. Inicia enfermedad actual en el mes de mayo de 2013 tras evidenciar dolor en la mucosa gingival de la mandíbula debido al aumento de volumen facial a predominio de hemicara izquierda. Dentro de los antecedentes personales encontramos: alergia al ácido acetilsalicílico, hipofunción tiroidea diagnosticada en el año de 1995, resección de tumor en fémur izquierdo, resección de tumor en húmero izquierdo con colocación de autoinjerto, biopsia de tejido óseo de cráneo, resección de tumor supraacetabular izquierdo con colocación de cemento óseo, afeitado maxilar. Al examen físico se evidencia discrepancia entre los tercios faciales, así como también en el análisis de los quintos faciales; esto se traduce en asimetría facial por aumento de volumen a expensas de hemicara izquierda (Fig. 1). A nivel ocular presenta proptosis marcada del ojo izquierdo, asimetría entre los pabellones auriculares en cuanto al ángulo de la concha con respecto a la mastoides siendo más evidente en el lado izquierdo, también presenta incompetencia labial, borramiento de surco nasogeniano y labiomenton bilateral (Fig. 1).
Fig.
1
Vista lateral derecha, detalle de la deformidad facial en región
cutánea del labio.
Vista frontal, detalle de la deformidad facial a predominio izquierdo.
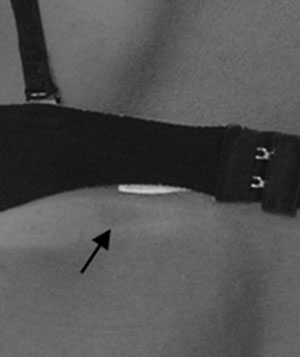
Ante el examen del esqueleto apendicular se denota simetría en ambos miembros superiores, hay presencia de escoliosis (Fig. 2) asociada al hecho de que el miembro inferior derecho es más largo que el izquierdo por lo cual se adopta una posición compensatoria (Fig. 2).
Fig. 2
Vista posterior, detalle de laescoliosis.
Discrepancia entre longitud
de miembros inferiores.
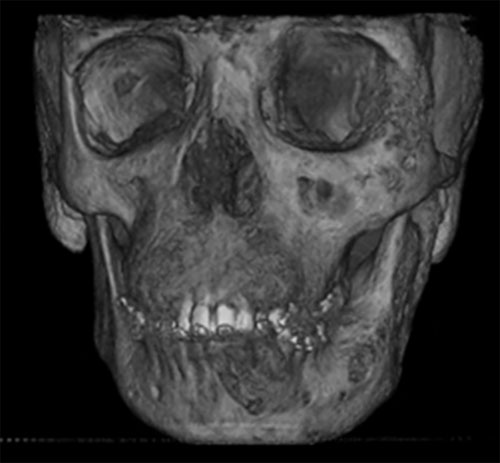
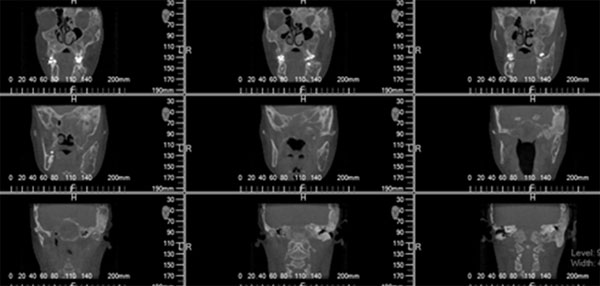
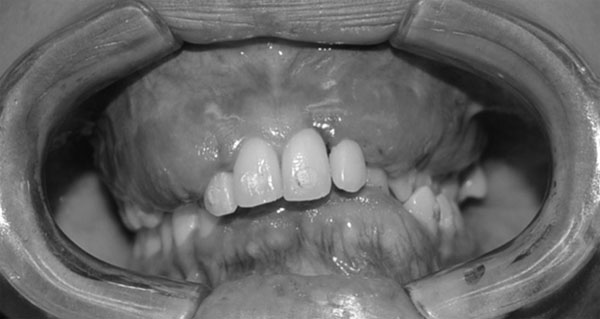
En la evaluación tomográfica de conebeam con reconstrucción 3D, se denota afectación de hemimandibula, hueso malar, pilar frontomalar, base de cráneo a nivel de fosa craneal anterior, media y posterior del lado izquierdo (Fig. 3). En el examen clínico intrabucal se encuentra crecimiento óseo bimaxilar deformante, múltiples diastemas (Fig. 4).
Fig. 3
Reconstrucción 3D Corte coronal Fig. 4
Foto intrabucal
Las manchas café con leche se encontraban en la región malar, preauricular, maseterina izquierda y en región infraescapular izquierda (Fig. 5). Existe concordancia entre el sitio de afectación de la FD y las manchas café con leche ya que estos dos signos afectaron al hemicuerpo izquierdo. La paciente refiere que con el tiempo las machas fueron atenuándose hasta desaparecer algunas quedando solo en zonas muy puntuales.
Fig.
5
Vista posterior, detalle de macula cutánea café con leche en región
infra escapular izquierda
Vista
lateral izquierda, detalle de maculas cutáneas café con leche en
región malar y maseterina izquierda.
Presentó metrorrágia precoz único a los 9 meses de edad el cual se repitió a los 5 años y persistió de manera intermitente, con duración de 3 días y de forma irregular cada 2-3 meses. A los 20 años de edad fue diagnosticada con ovarios poliquísticos por lo cual se le prescribió etinilestradiol y acetato de ciproterona lo cual reguló el ciclo menstrual. Según paciente los caracteres sexuales secundarios se manifestaron a los 4 años con la aparición de los botones mamarios; posteriormente a los 8 años de edad apareció el vello púbico y axilar. Como endocrinopatía asociada, la paciente cursa con hipotiroidismo actualmente en tratamiento.
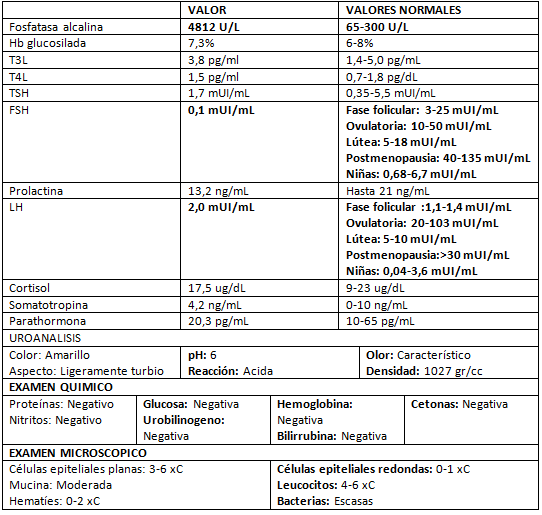
Se indicaron varios exámenes de laboratorio para determinar el perfil endocrinológico actual de la paciente (Tabla I) en los cuales los únicos valores alterados fueron la fosfatasa alcalina con valores muy por encima de lo normal, lo cual coincide con su condición la cual es la de una FD poliostótica, las hormonas folículo estimulante y luteinizante muy por debajo de los valores normales de una niña.
Tabla I:
Exámenes de laboratorio
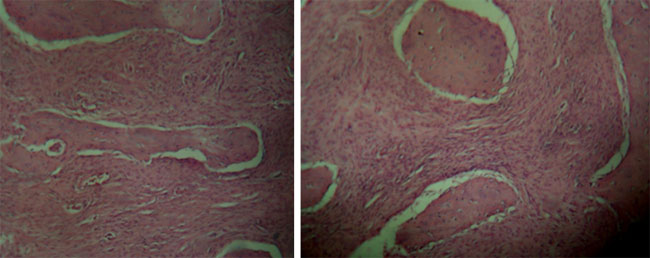
Se le realiza intervención el día 18/07/2013 la cual consistió en toma de biopsia y afeitado de región mandibular izquierda ordenándosele el estudio histopatológico. En el estudio macroscópico de la muestra a analizar se describen 3 fragmentos irregulares, dos blandos y uno duro, de color negro, midiendo en conjunto 2x1x0,5 cm. En el micro se evidenció estroma de tejido conectivo fibroso denso, con áreas mixoides, en el que se identifican, trabéculas de tejido óseo, inmaduro y maduro vital, de forma y tamaño variado, sin ribete de osteoblastos, algunas conectadas entre sí. Se identifican vasos sanguíneos de variable calibre, algunos hiperhémicos. Se trata de una lesión benigna denominada displasia fibrosa (Fig. 6).
Fig. 6
Microfotografías, se evidencio estroma de tejido conectivo fibroso denso, con áreas mixoides, en el que se identifica, trabeculas de tejido óseo, inmaduro y maduro vital. Cortesía del centro JUMA CA; centro de especialidades odontológicas. DISCUSIÓN
El diagnostico del SAMS clásicamente consiste en la triada de pubertad precoz, maculas cutáneas café con leche y FD ósea. La mutación y activación de la subunidad alfa de la proteína G es la anormalidad genética responsable del síndrome. El SAMS puede tener manifestaciones clínicas heterogéneas dependiendo del número de células mutadas y órganos afectados. En la literatura concerniente al SAMS indican que la mayoría de los casos reportados de fueron presentados como la forma atípica o incompleta del síndrome y presentaron casos raros que cursaron con los síntomas clásicos. Parece que una definición más práctica del SAMS podría ser cualquier posible combinación de FD con alguna de las otras dos características clínicas, pubertad precoz maculas cutáneas café con leche 2.
En el estudio en cuestión se reporta un caso de SAMS clásico el cual tuvo como primera manifestación la metrorragia asociado a aparición temprana de caracteres sexuales secundarios, con botones mamarios a los 4 años, vello púbico y axilar a los 8 años; todos estos signos indican la presencia de PP, ya que la normalidad indica que los límites normales de inicio de pubertad se mantienen entre los 9 y 11 años en niñas blancas europeas (media 10,7 años) y el final de ella a los 15,2 años. Aunque se ha observado una tendencia secular al adelanto puberal, parece haberse estabilizado desde los años ochenta. La edad media de menarquía es los 12,5 años en niñas blancas (rango 10-15 años) 13.
Las machas café con leche estuvieron presentes desde el nacimiento siendo estas dos características clínicas confirmatorias del SAMS, la aparición posterior de la FD hace que se redescriba como un síndrome y se inician los estudios pertinentes, se diagnostica el SAMS con las características clínicas de pubertad precoz, machas café con leche y FD ósea poliostótica2.
La FD ósea es una condición benigna en la que el hueso normal y medula ósea son reemplazados por tejido fibroso 14,13. La asimetría e inflamación son las quejas más comunes cuando la FD se encuentra en huesos faciales. Las deformidades secundarias debidas a crecimiento lento de FD incluyen distopía vertical, proptosis, canteamiento o asimetría facial y de la mandíbula. El grado de deformidad varia pero aquellos con SAMS más severamente afectados. Las lesiones de FD faciales pueden ser descritas como estacionarias, no agresivas de crecimiento lento, o agresivas de crecimiento rápido con dolor y parestesia, fracturas patológicas, transformación maligna, asociación a lesión secundaria 7. En concordancia con el caso presentado en el cual se presenta un caso típico de FD ósea poliostótica, la paciente no presentó transformación maligna pero si las características faciales descritas con anterioridad y el rápido crecimiento con dolor y parestesia asociado. Como diagnóstico diferencial se debe tener en cuenta el síndrome de Jaffé Lichstenstein el cual cursa con múltiples focos de quistes óseos aislados de FD ósea poliostótica, aparece en la infancia y adolescencia, en contraste con el SAMS donde la FD se asocia a otros signos cardinales confirmatorios del síndrome 16.
Las manchas café con leche cuando están presentes suelen ser la primera manifestación del SAMS, usualmente apareciendo al nacimiento o poco tiempo después de este. Pueden ser una pista temprana para el diagnóstico. Se describen clásicamente como manchas café con leche con bordes irregulares (en costa de Maine) a diferencia de las manchas vistas en la neurofibromatosis las cuales presentan un borde regulares (en costa de California). Estas manchas usualmente no tienden a cruzar la línea media y no se ha demostrado correlación entre el sitio de aparición de las manchas con el sitio de afectación de la FD ósea 17. En el caso presentado las manchas café con leche fueron más evidentes en la infancia, estas fueron desapareciendo con los años, disminuyendo su intensidad dejando solamente manchas puntuales en las zonas antes descritas, en la literatura revisada no hay mención sobre la desaparición de las manchas o su atenuación en cuanto la intensidad de su color.
La metrorragia como primer síntoma relacionado al SAMS, no fue tratado tempranamente ni directamente y no tuvo relevancia para los padres, sino hasta los 20 años de edad cuando se diagnostica con ovarios poliquísticos y comienza a ser tratada con etinilestradiol y acetato de ciproterona; a raíz de esto se consiguió regular el ciclo menstrual. Algunos autores describen que el tratamiento ideal para la pubertad precoz no es claro. La medroxiprogesterona y acetato de ciproterona pueden ser efectivos para aliviar la metrorragia, pero no tienen efecto en la edad ósea 18,19. A pesar de haber sido diagnosticada con el SAMS desde el año 1999 donde tenía 8 años de edad.
En la revisión de la literatura se puede apreciar que una endocrinopatía que puede estar presente en dicho síndrome es el hipertiroidismo. Este último puede aparecer de manera subclínica o hipertiroidismo franco con bocio, este se encuentra precedido por la pubertad precoz en cuanto a frecuencia de aparición 20. En el caso presentado se determina que este aparece como hipotiroidismo desde el año 1995 siendo tratado con levotiroxina hasta la actualidad. La paciente no presenta valores de hormona de crecimiento aumentada ni fenotipo que indique su aumento.
La FD es la característica clínica que hace que el paciente acuda a la consulta odontológica, esta afecta huesos largos y huesos craneofaciales, en los estadios iníciales del SAMS el paciente debe consultar con un equipo multidisciplinario dentro del cual se destaca la participación del odontólogo y cirujano buco-maxilofacial en lo concerniente a lesiones bucales asociadas a la FD. Por esto se debe tener un conocimiento claro de los signos cardinales del SAMS para así poder diagnosticar correctamente el mismo y orientar al paciente en cuanto a tratamiento se refiere.
REFERENCIAS BIBLIOGRÁFICAS
Classen CF, Mix M, Kyank U, Hauenstein C, Haffner D. Pamidronic acid and cabergoline as effective long-term therapy in a 12-year-old girl with extended facial polyostotic fibrous dysplasia, prolactinoma and acromegaly in McCune-Albright syndrome: a case report. J Med Case Rep. 2012 Jan 24;6:32. doi:10.1186/1752-1947-6-32. PubMed PMID: 22273876; PubMed Central PMCID: PMC3277484.
Rostampour N, Hashemipour M, Kelishadi R, Hovsepian S, Hekmatnia A. A Case of Atypical McCune-Albright Syndrome with Vaginal Bleeding. Iran J Pediatr. 2011 Sep;21(3):399-403. PubMed PMID: 23056821; PubMed Central PMCID: PMC3446187.
Zhou J, Sun LH, Cui B, Song HD, Li XY, Ning G, Liu JM. Genetic diagnosis of multiple affected tissues in a patient with McCune-Albright syndrome. Endocrine.2007 Apr;31(2):212-7. PubMed PMID: 17873334.
Akintoye SO, Lee JS, Feimster T, Booher S, Brahim J, Kingman A, Riminucci M, Robey PG, Collins MT. Dental characteristics of fibrous dysplasia and McCune-Albright syndrome. Oral Surg Oral Med Oral Pathol Oral RadiolEndod. 2003 Sep;96(3):275-82. PubMed PMID: 12973283.
Lee JS, FitzGibbon EJ, Chen YR, Kim HJ, Lustig LR, Akintoye SO, Collins MT, Kaban LB. Clinical guidelines for the management of craniofacial fibrous dysplasia. Orphanet J Rare Dis. 2012 May 24; 7Suppl 1:S2. Doi: 10.1186/1750-1172-7-S1-S2. Epub 2012 May 24. PMCID: PMC3359960.
Patel KB. McCune-Albright syndrome: a case report in a male. Indian J DermatolVenereolLeprol. 2010 Nov-Dec;76(6):723. doi: 10.4103/0378-6323.72473. PubMed PMID: 21079331.
Ponti G, Tomasi A, Pastorino L, Ruini C, Guarneri C, Mandel VD, SeidenariS,Pellacani G. Diagnostic and pathogenetic role of café-au-lait macules in nevoid basal cell carcinoma syndrome. Hered Cancer ClinPract. 2012 Oct 29;10(1):15. doi: 10.1186/1897-4287-10-15. PubMed PMID: 23107377; PubMed Central PMCID:PMC3502463.
Kim IS, Kim ER, Nam HJ et al. Activating mutation of GS alpha in McCune-Albright syndrome causes skin pigmentation by tirosinase gene activation on affected melanocytes. Horm Res, 52:235-40, 1999.
Precocious puberty in girls. Indian J EndocrinolMetab. 2012 December; 16 (Suppl 2): S188-S191. doi: 10.4103/2230-8210.104036 PMCID: PMC3603023.
Hernández L, Espinosa AL, Méndez V, Nishimura E, Mercado M. Síndrome de McCune-Albright: características clínicas en una población pediátrica y adulta. Revista de Endocrinología y Nutrición, 2012 Enero-Marzo Vol. 20, (1)pp 11-8.
Riminucci M, Fisher LW, Shenker A, Spiegel AM, Bianco P, GehronRobey P: Fibrous dysplasia of bone in the McCune-Albright syndrome: abnormalities in bone formation. The American journal of pathology 1997, 151(6):1587-1600. PMCID: PMC1858361.
Riminucci M, Liu B, Corsi A, Shenker A, Spiegel AM, Robey PG, Bianco P. The histopathology of fibrous dysplasia of bone in patients with activating mutations of the Gs alpha gene: site-specific patterns and recurrent histological hallmarks. J Pathol. 1999 Jan;187(2):249-58. PubMed PMID: 10365102.
Peltier J, Lefranc M, Fichten A, Cordonnier C, Toussaint P, Desenclos C, Le Gars D. Odontoid fracture complicating Jaffe-Lichtenstein disease. Case report. J Neurosurg Spine. 2008 Mar;8(3):295-9. doi: 10.3171/SPI/2008/8/3/295. PubMed PMID:18312084.
Rao S, Colaco MP, Desai MP. McCune Albright Syndrome (MCAS): a case series. Indian Pediatr. 2003 Jan;40(1):29-35. PubMed PMID: 12554915.
Sorgo W, Kiraly E, Homoki J, Heinze E, Teller WM, Bierich JR, Moeller H, Ranke MB, Butenandt O, Knorr D. The effects of cyproterone acetate on statural growth in children with precocious puberty. ActaEndocrinol (Copenh). 1987 May;115(1):44-56. PubMed PMID: 2954358.
Shenker A, Weinstein LS, Sweet DE, Spiegel AM. An activating Gs alpha mutation is present in fibrous dysplasia of bone in the McCune-Albright syndrome. J ClinEndocrinolMetab. 1994 Sep;79(3):750-5. PubMed PMID: 8077356.
Collins MT, Singer FR, Eugster E. McCune-Albright syndrome and the extraskeletal manifestations of fibrous dysplasia. Orphanet J Rare Dis. 2012 May 24;7Suppl 1:S4. doi: 10.1186/1750-1172-7-S1-S4. Epub 2012 May 24. Review. PubMed PMID: 22640971; PubMed Central PMCID: PMC3359955.