Cirugía Ortognática en paciente con Ostegénesis imperfecta, presentación de un caso y revisión de la literatura
Recibido para arbitraje: 06/07/2009
Aceptado para publicación: 01/12/2009
MEDINA ESQUIVEL MARIA TERESA* LICÉAGA REYES RODRIGO§,
* Residente de 4to año, Servicio de Cirugía Oral y Maxilofacial, Hospital Juárez de México.
§ Médico Adscrito al Servicio de Cirugía Oral y Maxilofacial, Hospital Juárez de México.
Resumen
La osteogénesis imperfecta es una enfermedad genética que afecta la calidad de la colágena tipo 1, por lo que la matriz ósea y otros tejidos conectivos se ven afectados en distintos grados. La penetrancia de esta enfermedad es muy variable, y puede ser desde leve hasta incompatible con la vida.
Presentamos la revisión de la literatura en conjunto con el caso de una paciente de 32 años de edad con diagnóstico de osteogénesis imperfecta que fue sometida a una mentoplastía como tratamiento de cirugía ortognática.
Abstract
Ostegenesis imprefecta is a genetic disease which affects the quality of collagen type 1, so osseous matrix and other connective tissues are affected in different degrees. The severity of the disease could be mild or so severe that is not possible to live with it.
We present a review of the literature and report the case of a 32-year old female patient with osteogenesis imperfecta who went under a genioplasty as a procedure or orthognatic surgery.
Key Words: Osteogenesis Imperfecta, Genioplasty.
Introducción
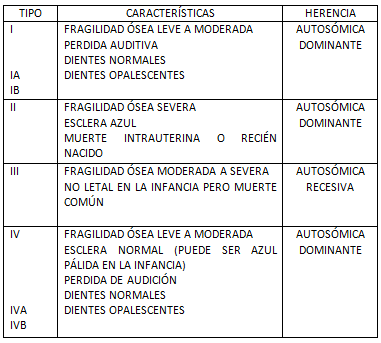
Osteogénesis imperfecta (OI) es un grupo heterogéneo de enfermedades hereditarias de del metabolismo de la colágena tipo I caracterizada por fragilidad ósea. Algunas características asociadas pueden incluir esclera azul, perdida de audición, deformidad de huesos largos y vértebras e hipermovilidad articular (1).
El primer caso reportado fue de un egipcio que data del año 1000 AC que se encuentra en el Museo Británico (2). Ekman realizo el primer estudio completo de este síndrome en 1978 donde implico su carácter hereditario (3). Lobstein (3) y Vrolik (4), en 1900, explicaron las características de la enfermedad tanto en el adulto y en el recién nacido respectivamente. Es por eso que a la fecha se le conoce a la tipo I como enfermedad de Lobstein y a la tipo II como enfermedad de Vrolik.
Van der Hoeve y Kleijn mencionaron la sordera como parte del síndrome . Preiswerk fue el primero en describir las anormalidades dentales. La clasificación clínica actual fue descrita por Sillence (6).
La variabilidad de la enfermedad es muy grande, incluso entre miembros de la misma familia. Mientras algunos individuos pueden tener afecciones mínimas y nunca tener una fractura, otros pueden padecerlas desde el nacimiento (7).
La prevalencia estimada de todo este grupo es de 1 en cada 20,000 nacimientos. No hay una predilección por sexo ni por un tipo racial.
Las distintas causas de osteogénesis imperfecta son ocasionadas por mutaciones en los dos genes que codifican para colágena tipo I (COL1A1 del cromosoma 17, y COL1A2 del cromosoma 7). Estos genes codifican para la pro-?, que son cadenas de procolágena, la cual es secretada por los fibroblastos. Si esta formación no es uniforme, las estructuras de colágena tendrán estructura anormal (8,9).
Las características generales cambian entre los distintos tipos de severidad de la enfermedad.
No existe una fascies característica excepto por la esclera azul, aunque la OI tipo I ha sido frecuentemente descrita con hipoplasia maxilar y prognatismo mandibular relativo (10).
En todos los tipos de OI se pueden presentar también problemas cardiovasculares de diferente pronóstico.
A pesar de la deformación ósea y la frecuencia de fracturas, la longevidad de una persona afectada por OI es igual a la de cualquier otra. Sus cualidades intelectuales no están mermadas de ninguna forma por la enfermedad y pueden llevar una vida normal.
Los tres síntomas clásicos de la OI incluye fragilidad ósea, pérdida temprana de la audición y escleróticas con apariencia azulosa (escleróticas azules), sin embargo, no todas las personas afectadas por la OI tienen escleróticas azules o pérdida de la audición. Todos sufren de fragilidad ósea, pero no todos sufren de fracturas en los huesos (11).
Características que podemos encontrar en los pacientes con OI:
Fractura ósea
Fractura múltiple en un sólo episodio
Fracturas congénitas
Por trauma menor
Deformidad de las extremidades
Sordera
Cifosis
Cifoescoliosis
Baja estatura
Anormalidades dentales
Puente nasal bajo
Pectus carinatum
Pectus excavatum
Pes planus
Laxitud de las articulaciones
Tendencia a la formación de hematomas
Piernas en arco
Un examen físico puede confirmar la presencia de fracturas, deformidades y otros síntomas. Las radiografías pueden mostrar fracturas múltiples que han sanado. El diagnóstico definitivo El diagnóstico se confirma al medir la producción de procolágena tipo I por fibroblastos dérmicos en cultivo. Una vez conocido el diagnóstico molecular específico, a los miembros de la familia se les puede hacer una prueba por medio de un examen de sangre para ADN.
Al ser un padecimiento de la colágena el tratamiento de la OI no es administrando calcio complementario. No existe hasta el presente ninguna forma de inducir a las células del cuerpo a producir más colágeno o producir colágeno de calidad. Experimentalmente, se ha propuesto la administración de bifosfonatos con objeto de reducir la pérdida ósea.
Reporte del caso
Paciente femenino de 32 años de edad, con antecedente traumático de fracturas espontáneas de parietal, de fémur derecho a los 3 años, y de codo bilateral a los 6 años de edad. Padece importante fragilidad capilar lo que en edades tempranas aparentaba violencia interfamiliar hasta que a la edad de 11 años se realizo el diagnóstico de osteogénesis imperfecta tipo IA (OI) con mutación de novo.
Fig. 1
Fotografía preoperatoria. Exceso de proyección del mentón.
A los 16 años de edad busco tratamiento por tener perfil prognático y por la enfermedad de base el ortodoncista optó por realizar un camuflaje ortodóncico sin modificar el perfil facial (Fig. 1). Aunque se lograron buenos resultados dentales la paciente quedo insatisfecha ante el poco cambio facial, por lo que años después decidió ser revalorada. Por dolor dental se le extrajeron los terceros molares, y durante el transoperatorio se encontró hueso aparentemente normal que cicatrizó de manera convencional sin complicaciones, consideración que posteriormente fue de mucha importancia para la toma de decisiones de intervenir el mentón. Esto animó a la paciente a intervenirse el mentón.
Se realizaron los estudios cefalométricos de tejidos duros y blandos correspondientes, y al correlacionarlos con las medidas clínicas y las fotografías intra y extraorales obtenidas (Fig. 1) para integral el diagnóstico.
El La conclusión de los estudios demostró una disarmonía dentofacial por un exceso de crecimiento del cuerpo mandibular. Presentaba adecuada exposición dentogingival, y debido a la oclusión que ya se había logrado ortodóncicamente se optó por únicamente trabajar sobre el mentón con una mentoplastía deslizante de retroceso con disminución de la dimensión vertical del mismo.
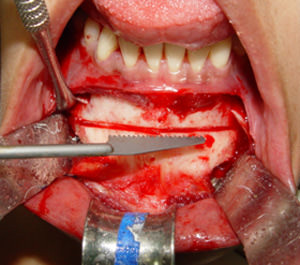
Fig. 2
Aspecto transoperatorio de la osteotomía.
Una vez acordado el procedimiento quirúrgico a realizar se obtuvo bajo consentimiento informado la autorización escrita del paciente.
En los estudios preoperatorios de rutina no se encontró anormalidad alguna, incluidos estudios de perfil tiroideo y CPK total, por lo que la valoración preoperatoria ni tuvo consideraciones especiales para el caso.
La cirugía se realizo en quirófano bajo anestesia general balanceada con intubación nasotraqueal, con un abordaje intraoral en fondo de saco vestibular, exponiendo toda la zona del mentón y cuidando de la integridad de ambos nervios mentonianos.
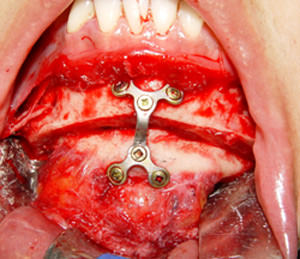
Con una sierra reciprocante se realizo la osteotomía deslizante (Fig. 2) para que una vez completado con cinceles se recolocara en su nueva posición por medio de una placa de titanio y 6 tornillos monocorticales de 2.0 por 7 mm (Fig. 3). El movimiento final fue un retroceso de 5 mm y 6 mm de reducción vertical. El hueso encontrado tenía características macroscópicas normales, con vascularidad aparentemente normal. La porción de hueso eliminada se envió a estudio histopatológico.
Fig. 3
Fijación interna
Se suturo por planos con sutura absorbible (vicryl 3-0) y se coloco una mentonera que facilitara la reinserción muscular y prevenir la aparición de hematomas o seromas.
Se administraron antibióticos, analgésicos y antiinflamatorios esteroideos de manera convencional y fue dada de alta al día siguiente para vigilancia por consulta externa.
Se mantuvo en citas semanales durante el primer mes y después cada tres meses a conveniencia del equipo quirúrgico.
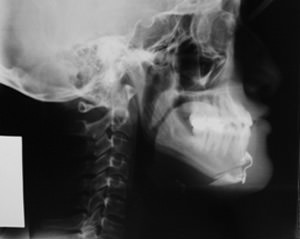
Fig. 4
Radiografía Postoperatoria.
Los controles radiográficos mostraron una adecuada unión de los segmentos óseos (Fig. 4).
La paciente obtuvo el cambio facial esperado por medio de este procedimiento sin someterse a un tratamiento mayor como lo sería un retratamiento de ortodoncia y una cirugía ortognática más compleja (Fig. 5).
Fig. 5
Fotografía posoperatoria. Discusión
Es de vital importancia para el clínico saber que la OI tipo I es de alrededor de 1 por cada 30 mil habitantes (12,13) por lo que tiene que ser muy cuidadoso en el abordaje ante la sospecha de dicha enfermedad.
Sin embargo, en la actualidad y de acuerdo a los avances en medicina la gran mayoría de estos casos son diagnosticados a temprana edad por lo tanto llegarán con un diagnóstico ya establecido.
Las consideraciones específicas a la cirugía maxilofacial en pacientes con OI están relacionadas a la: 1) densidad ósea y la propensión a las fracturas, 2)la posibilidad de no unión secundaria a una alteración microvascular del periostio, 3)sangrado causado por desordenes plaquetarios y defectos endoteliales microvasculares, y 4)complicaciones anestésicas, incluyendo la hipertermia maligna.
En relación al inciso uno, Falvo y cols, describen la histología básica de hueso afectado de pacientes con OI obtenido por biopsia de cresta iliaca, el cual se caracteriza por grandes trabéculas óseas de patrón entrelazado típico (14), así mismo, algunos hallazgos radiográficos frecuentes son fracturas y lesiones óseas radiolúcidas (15,16,17) que sugieren osteopenia generalizada u osteoporosis. La cortical es delgada o no existente, que lo hace susceptible a fracturas, y pueden presentar quistes óseos idiopáticos. Algunas veces la formación de callo óseo resulta prominente y puede mostrar radiopacidad que se confunde con neoplasias (11,18,19,20). La formación de callo hiperplásico puede deberse a la calcificación del hematoma (21), de acuerdo a algunas hipótesis que puede semejar un sarcoma osteogénico, en un periodo de semanas a meses (11,18,20), . En sospechas de callo óseo, la fosfatasa alcalina (FA) y la velocidad de sedimentación globular (VSG) suelen estar elevados mientras que el calcio sérico y fósforo son normales (20,22).
Los callos hiperplásicos pueden hacerse evidentes durante la cicatrización ósea, sin embargo hay que tomar en cuenta que la mayoría de reportes de casos clínicos tienen un seguimiento a corto - mediano plazo, por lo que no se pueden concluir las consecuencias que puede tener este factor en los procedimientos realizados (11,18,23,24,25).
La no unión de fracturas en huesos largos ha sido bien documentada, pero no ha presentado un problema en la cirugía maxilofacial . Es importante tener cuidado al realizar las osteotomías para alinear adecuadamente los segmentos óseos hueso - hueso o usando injerto óseo interposicional con una adecuada fijación interna que optimice la cicatrización.
El reporte histopatológico que se obtuvo de la muestra obtenida durante la cirugía fue: fragmentos de hueso lamelar compacto vital con espacios medulares levemente esclerosados y ocupados por tejido adiposo maduro y tejido fibroso laxo, libres de alteración.
De acuerdo a la literatura de manera comparativa con el presente caso en donde no tuvimos complicaciones hemorrágicas graves podemos citar lo referido por Freedus y cols. quienes tampoco reportaron hemorragia grave en su reporte de caso de cirugía bimaxilar18, esto a consideración del 10% reportado por Hathaway y cols. en lo referente a sangrado excesivo por incremento de fragilidad capilar (27).
Se debe tener consideración que las pruebas de coagulación preoperatorias pueden no mostrar predicción cualitativa o cuantitativa de sangrado transoperatorio; pero siempre deben tomarse en todo paciente: tiempo de protrombina (TP), tiempo parcial de tromboplastina (TPT), fibrinógeno, plaquetas, tiempo de sangrado e índice internacional estandarizado (INR).
Por último se tuvo un especial cuidado en la posibilidad de la aparición de un aumento en la temperatura corporal del paciente durante el transoperatorio por parte del equipo de anestesiología, ya que de acuerdo con Cropp y Myers reportan hasta un 45% de incremento en el metabolismo, especialmente en niños (28). Así mismo se documenta hasta un 50% de elevación de hormonas tiroideas (28,29) lo que puede favorecer la aparición de hipertermia maligna en pacientes sometidos a anestesia general (29,30,31), así que dentro de las consideraciones específicas se encuentra el evitar agentes halogenados, relajantes musculares del tipo de la succinilcolina, anticolinérgicos (atropina), como posibles desencadenantes de hipertermia maligna. Consideramos importante mencionar que no fue posible determinar los niveles séricos y urinarios de pirofosfato inorgánico indispensable como predictor en la aparición de hipertermia maligna, ya que así lo demuestran los estudios de Solomons y Styner (32), y el de Armstrong y cols.(33) debido a que en nuestro medio no se cuenta con los recursos para dichas muestras.
Por otro lado la función plaquetaria depende del metabolismo de fosfatos los cuales pueden verse alterados en pacientes con OI lo que puede representar una alteración en la adhesión y retracción del coágulo importantes factores en caso de cirugía (34,35,36), así que de acuerdo a Tan y cols. (27) se tomaron muestras de CPK sérico y hormona tiroideas como parte del monitoreo.
Algunos reportes en la literatura y en relación al presente caso se ha documentado que aproximadamente el 75% de paciente con OI muestran maloclusión clase-III (38,39,40). Así mismo como es de considerar en el estudio reportado por O'Connell y Marini, hacen referencia a que no se contraindica el uso de tratamiento ortodóncico como posible factor de riesgo de lesión dentaria38, y así lo demostró el antecedente de historia clínica de la paciente.
Conclusiones
El presente caso muestra las consideraciones a tomar en cuenta en casos no graves de osteogénesis imperfecta a la decisión de realizar cirugía ortognática con bajas posibilidades de complicaciones.
Referencias
Gray PHK: A case of osteogenesis imperfecta associated with dentinogenesis imperfecta, dating from antiquity. Cli radiol 1969; 20:106-108.
Ekman OS: Descriptionem et casus aliquot osteomalacies sistens: Disertartio Medica Uppsala, Sweden, 1788, citado en Weil UV: osteogenesis imperfecta historical background Clin Orthop Rel Res 1981; 159:611.
Lobstein JFGCM: De la fragilité des os, ou de de l'ostepsathyrose. Traite de l'Anatomie Patologique, Vol 2, FG Lerrault, Paris, 1833, 99-204-212, citado en Weil UV: osteogenesis imperfecta: historical background. Clin Orthop Rel Res 1981; 159:6-11.
Vrolik W: Tabulae ad illustandum embrygenesim hominis et malium, tam naturatem quam abnomen, Amsterdam, 1849. Citado en Weil UV: osteogenesis imperfecta: historical Clin Orthop Rel Res 1981; 159:6-11.
Van der Hoeve J, de Klejn A: Blau Scleae, Knochenbruchigkeit und Schwerhorigkeit. Arch ophthalmol 1918; 95:91-93.
Silence DO: Genetic heterogeneity in osteogenesis imperfecta. J Med Genet 1979;16:101-116.
Stoll C Birth prevalence rates of skeletal displasias. Clin Genet 1989; 35:88-9
Byers PH. Osteogenesis imperfecta: translation of mutation to phenotype. J Med Genet 1991; 28: 433-442.
Prockop DJ: Mutations that alter the primary structure of type I collagen. J Biol Chem 1990; 265: 15349-15352.
Cole NL, Goldberg MH, Loftus M, Kwok V. Surgical management of patients with osteogenesis imperfecta. J Oral Maxilofac Surg 1982; 40:578-584.
Bawle EV: Osteogenesis imperfecta vs. child abuse. Am J Med Genet 49:131,1994.
Kasper DL, Fauci AS, Longo DL, Braunwald E, Hauser SL, Jameson JL. Harrison`s Principles of Internal Medicine , 16 th ed, McGraw-Hill, 2005, p 2557-2566.
Falvo KA, Root L, Bullough PG. osteogenesis imperfect: A histometric analysis. J Bone joint Surg 1973;55:275-286.
Levin LS, Weigth JM, Byrd DL, Greenway G, Dorst JP, Irani RN. Osteogenesis imperfecta with unusual skeletal lesions: Report of three families. Am j Med Genet 1985; 21:257-259.
Bergstrom L. Osteogenesis imperfect Otologic and maxillofacial aspects. Laryngoscope 1997; 87:142.
Lukinmaa PL, Ranta H, Ranta K, Kaitila l, Hietanen J. Dental findings in osteogenesis imperfecta: II. Dysplastic and other devolpmental defects. J Craniofac Genet Dev Bio 1987;7:127-135.
Freedus MS Schaaf NG,Zitter WD: Orthognathic surgery in osteogenesis imperfecta. J Oral Surg 34:830-34, 1976.
Moorefield WG Jr, Miller GR: Aftermath of osteogenesis imperfect: the disease in adulthood J. Bone Joint Surgery 62:113, 1980.
Banta JV, Schreiber RR, Kulik WJ: Hyperplastic callus formation in osteogenesis imperfect simulating osteosarcoma. J Bone Joint Surg 53:115,1971.
Laurent LE, Salenius P: Hyperplastic callus formation in osteogenesis imperfecta. Acta Orthop Scap 38:280;1967.
Lewis MK, Stoker NG. Surgical management of the patient with osteogenesis imperfect, J Oral Maxillofac Surg 1987; 45:430-437.
Ornisiston IW, Tideman H. orthognatic surgery in osteogenesis imperfecta: A case report with management considerations. J craniomaxillofac Surg 1995, 23:261-265.
Eppley BL, Kalsbeck JE, Sabove AM. Cranial reconstruction in osteogenesis imperfecta. J Craniomaxillofac surg 1994;5;3:180-184.
Gamble JG, Rinsky LA, Strudwick J, Bleck EE. Non-union of fractures in children who have osteogenesis imperfecta. J Bone Joint Surg 1988;70:439-443.
Hathaway WE, Solomons CC, Ott JE. Platelet function and pyrophosphates in ostegenesis imperfecta Blood 1972;39:500-509.
Cropp GJ, Myers DN. Physiological evidence of hypermetabolism in osteogenesis imperfecta. Pediatrics 1972;49:375.
Solomons CC, Meyes DN. Hyperthermia of osteogenesis imperfecta and its relationship.
Solomons CC, Millar E. Osteogenesis imperfecta: New perspectives. Clin Orthop 96:299, 1973.
Depinna GA: A case of osteogenesis imperfecta malignant hyperthermia susceptible-anesthetic management. Second international symposium on malignant hyperthermia. New York, Grune and Stratton, 1978,p409.
Solomons CC, Styner J: Osteogenesis imperfecta: Effect of magnesium administration on pyrophosphate metabolism. Calcif tissue Res 3:318,1969.
Armstrong D, Van Wormer D, Solomons CC: increased inorganic serum pyrophosphate inn serum and urine of patients with osteogenesis imperfecta . clin Chem 21:104,1975.
Hathaway WE, Solomons CC, Ott JE: platelet function and pyrophosphates in osteogenesis imperfecta. Blood 1972;39:500-509.
Siegel BM, Friedman IA, Schwartz SO: Hemorrhagic disease in osteogenesis imperfecta. Am J Med 1957;22:315.
Hathaway WE, Solomons CC, Ott JE: Abnormalities of platelet function in osteogenesis imperfecta. Clin Res 1972; 18:209.
Tan S, Aldrete JA, Solomons CC: Correlation of serum creatinine phosphokinase and pyrophosphate during surgery in patients with malignant hyperthermia susceptibility second international symposium on malignant hyperthermia. New York, Grune and Stratton, 1978,p389.
O ' Connell AC, Marini JC. Evaluation of oral problems in an osteogenesis imperfecta population. Oral surg Oral Med Oral Pathol Oral Radiol Endod 1999;87,189-96.
Schwartz S, Tsipouras P, Oral findings in osteogenesis imperfecta. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1984;57;161-167.
Stenvik A, Larheim TA, Stohaug K. incisor and jaw relationship in 27 persons with osteogenesis imperfecta. Scand J Dent Res 1985; 93;56-60.